

서 론
재료 및 방법
식물재료 및 ABA 처리
RNA 추출 및 Illumina sequencing
Read mapping 및 RNA-seq 데이터 분석
결과 및 고찰
Illumina sequencing 분석 결과
Differentially expressed genes (DEGs)분석
ABA 생합성 경로 관련 유전자
서 론
밀은 세계적으로 옥수수, 벼 다음으로 많이 소비되는 식량작물이다(FAO 2013). 국내 밀 소비량은 지속적으로 늘어나는 추세이나 자급률은 1.1%에 불과한 실정이다. 겨울철 저온이나 생육기간 내의 한발 같은 수분 스트레스 등의 국내 재배환경 등이 주요한 문제로 판단되고 있다. 한편 국외에서도 밀에 대한 연구는 많이 수행되고 있지만 allohexaploid라는 복잡한 genome 구성으로 인하여 연구에 어려움이 많은 실정이다.
Abscisic acid (ABA)는 gibberellic acid (GA), cytokinins (CKs), ethylene (ET), auxin (IAA)와 함께 식물에서 발견되는 주요한 호르몬 중 하나이며 종자의 휴면, 발아 및 스트레스에 관련된 식물호르몬이다. 종자의 휴면 및 발아에서는 GA와 길항적으로 작용하는 것으로 알려져 있으며 (Holdsworth et al., 2008; Nambara et al., 2010; Nonogaki 2014), 또한 식물의 수분 스트레스 반응에 관여하여서는 한발 저항성에 중요한 역할을 하는 것으로 알려져 있다(Assmann 2003; Christmann et al., 2007; Tardieu, Parent et al., 2010).
지난 몇 년간 sequence의 대량분석 기술은 급진적으로 발전해왔으며 비용 또한 많이 감소하였다. RNA-seq은 유전자 발굴 및 다양한 스트레스에 대해 반응하는 유전자의 발현 분석에 매우 효과적인 기술로써 RNA-seq을 통해 많은 sequence data, 발현 profiling 정보가 공개되었으며 이는 model 식물이나 genome sequence가 공개된 식물 뿐만 아니라 genome sequence가 알려지지 않은 식물을 포함하여 non-model 식물에서도 많이 연구 및 분석이 수행되었다(Zhang et al., 2010; Kakumanu et al., 2012; Wang et al., 2010).
본 연구에서는 Illumina sequencing을 통한 RNA-seq 분석을 통해 유묘기 밀에서 ABA를 처리했을 시 식물체 내에서 발현되는 대량전사체들의 변화를 알아보고 또한 ABA 관련 유전자들에 대한 분석을 수행하였다.
재료 및 방법
식물재료 및 ABA 처리
‘우리밀 (Korea RDA accession no. IT172221)’과 ‘D-7 (본 연구진 육성 계통, Fleming*4 /3/ PIO 2580 // T83103 *2 / Hamlet)’의 F4 세대 밀 육성라인이 실험에 사용되었다. 발아 처리한 종자들을 암조건의 생육상에서 4°C 에 4주간 춘화처리 한 후, Hoagland 용액을 채운 Incu Tissue (72×72×22 mm; SPL Life Sciences, 경기도)로 옮겼다. 12시간의 광주기, 20°C/15°C (day/night)와 60% 상대습도로 제어된 조건으로 14일 간 성장시켰으며, 14일 후의 유묘기 밀의 뿌리는 100 µM의 ABA를 처리하였다. 처리가 끝난 식물체는 실험을 하기까지 -80°C에 보관하였다.
RNA 추출 및 Illumina sequencing
ABA 처리 시 식물체 내에서의 transcriptome 분석을 위하여 각 유묘기 식물체 샘플의 전체 RNA는 TRIzol (Invitrogen, MA, USA)을 이용하여 추출하였다. RNA-seq 기법을 위한 paired-end library는 Illumina TruSeq RNA Sample Preparation Kit v2 (catalog #RS-122-2001; Illumina, CA, USA)를 이용하여 제작하였으며, RNA-seq은 HiSeq2000 platform을 이용하여 수행되었다. Short read의 전처리 과정을 위하여 SolexaQA (Cox et al., 2010)에서 제공하는 DynamicTrim과 LengthSort 프로그램을 사용하였으며 phred score Q≥20, short read length ≥25 기준으로 선발하였다.
Read mapping 및 RNA-seq 데이터 분석
Illumina sequencing 및 quality control을 통해 얻은 clean read들은 Bowtie2 (Langmead and Salzberg 2012) 프로그램을 이용하여 URGI (http://wheat-urgi.versailles.inra.fr/)로 부터 다운받은 밀의 draft reference gene sequence (ta_IWGSC_MIPSv2.2_HighConf_CDS_2014Jul18.fa) (International Wheat Genome Sequencing Consortium 2014)에 mapping하여 유전자 발현량을 분석하였다. 데이터의 편차에 대한 normalization은 DESeq library를 이용하여 수행하였다(Anders and Huber 2010).
획득한 transcripts의 염기서열을 amino acid 서열로 번역한 후, BLASTP (e-value 1e-10)를 이용하여 URGI DB에서 제공하는 유전자의 기능 정보와 Phytozome (Goodstein et al., 2012) DB의 amino acid 서열의 비교를 통해 gene annotation을 수행하였으며 ABA 생합성 경로 관련 유전자는 BLAST (e-value 1e-10, identity 70 이상)를 이용하여 선발하였다.
샘플 간 차별적으로 발현하는 유전자(DEGs; Differentially expressed Genes)는 발현값이 2배 이상 차이가 나는 2 fold change와 FDR (False Discovery Rate)값 0.01 이하를 만족하는 두 방법을 동시에 사용하여 선발하였다. 본 연구에서는 log2 값이 1 이상이면 up-regulation, -1보다 작으면 down-regulation 되었다고 명명하였다.
선발된 DEGs의 기능을 분석하기 위해 DEGs의 염기서열과 GO (Gene Ontology) DB에서 제공하는 서열을 비교하여 GO 분석(Ashburner et al., 2000)을 수행하였다. GO 분석은 web-based 분석 툴인 AgriGO의 singular enrichment analysis (SEA)를 이용하여 cut-off value를 FDR값 0.05 이하로 설정하여 다중비교 검정을 보정하였다.
결과 및 고찰
Illumina sequencing 분석 결과
유묘기 밀의 ABA 처리 후의 전사체 발현 분석 비교를 위하여 control과 ABA 처리 샘플에 대한 전체 RNA를 추출한 후 Illumina sequencing을 수행하였다. Control과 ABA 처리 샘플에서 각각 33,728,804개와 34,831,798개의 raw read들을 획득하였으며 이에 대한 quality control 등 pre-processing을 수행한 후 clean read들을 99,386개의 loci를 포함하고 있는 Triticum aestivum reference gene sequence에 mapping하였다. Control 샘플은 23,904,459개의 reads가 mapping 되었으며(77.07%), ABA 처리 샘플은 23,100,995개의 reads가 mapping 되었다(72.01%) (Table 1).

Differentially expressed genes (DEGs)분석
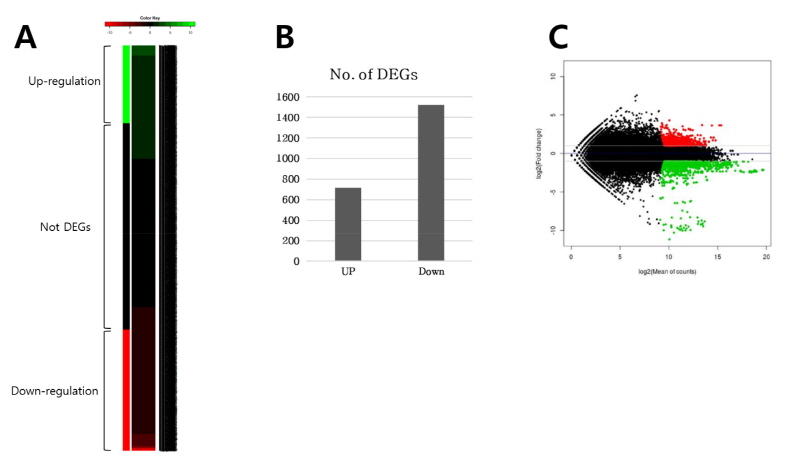
Reference gene sequence에 매치된 유전자들 중 raw read counts (>150)와 FDR 값 (<0.01) 이하를 만족시키는 유전자 총 5119개를 선발하였으며 이들에 대한 발현 패턴을 heatmap을 통해 분석하여(Fig. 1A) DEGs (2-fold change)를 확인하였다. Control 샘플과 비교해서 ABA 처리한 유묘기 밀에서 2,238개의 DEGs를 확인하였으며, (718개의 up-regulated와 1,520개의 down-regulated DEGs, Fig. 1B) 그 중 annotation된 DEGs는 up- 과 down- regulated 된 것이 각각 710개와 1,503개였다. DEGs의 발현량은 log2 발현값에 대한 fold change로 표현하여 도식화하였다(Fig. 1C). 식물체에 ABA 처리를 한 경우 한발과 비슷한 수분결핍 증상을 일으킨다고 잘 알려져 있으며, 광합성은 한발 스트레스에 굉장히 예민하여 net photosynthetic rate와 탄소고정 대사를 억제한다(Teng et al., 2014; Pinheiro and Chaves 2011). Up-regulation된 유전자들과 비교하여 볼 때, down-regulation된 유전자들 중 가장 많은 비율을 차지하는 것은 RuBisCo 및 광계(photosystem I, II)를 포함한 광합성 관련 유전자들이었다. 특히 RuBisCo 유전자는 94개가 down-regulation된 것으로 확인되며 광계 관련 유전자들은 I, II를 합쳐 약 120개 가량이 확인되었다. 엽록체 관련 유전자 또한 약 20개 가량이 포함되어 있었다.
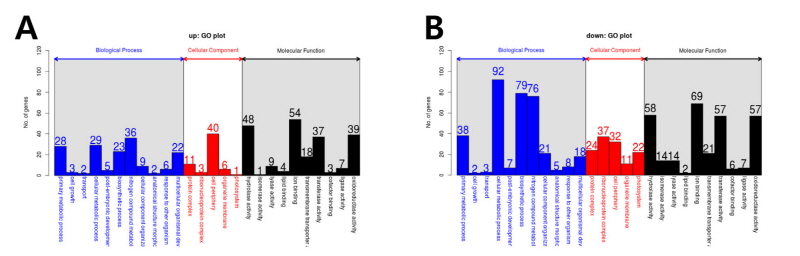
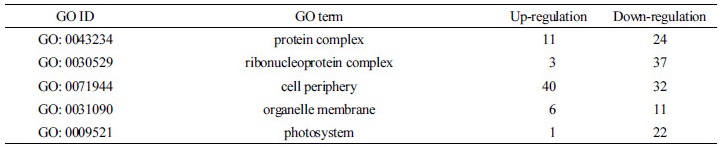
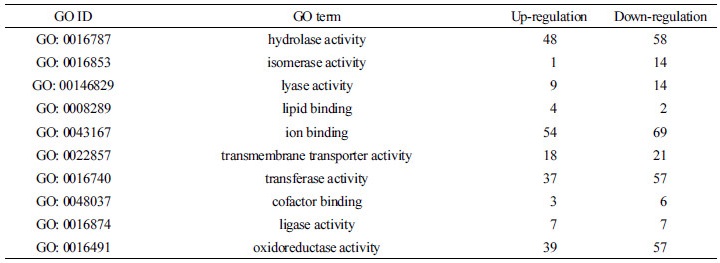
DEGs로 선발된 유전자들의 기능적 분류를 위해 GO 분석을 수행하였다. GO term은 크게 세 개의 주된 category (biological process, cellular component, and molecular function)로 나눌 수 있으며 이는 다시 26개의 sub-category로 나눌 수 있다. ABA 처리 시 확인된 2,238개의 DEGs 중 총 1,226개의 DEGs가 GO annotation 되었으며 biological process (BP)의 하위로는 cellular metabolic process가 약 23.5%로 가장 많은 비중을 차지는 그룹이었고 그 뒤로 nitrogen compound metabolic process (21.8%), biosynthetic process (19.8%), primary metabolic process (12.8%)가 순서대로 차지하였다. Cellular component (CC)에서는 cell periphery (38.5%), ribonucleoprotein complex (21.4%), protein complex (18.7%), photosystem (12.3%), organelle membrane (9.1%) 순이었으며, molecular function (MF)에서는 ion binding이 23.4%로 가장 큰 그룹이었고 뒤이어 hydrolase activity (20.2%), oxidoreductase activity (18.3%), transferase activity (17.9%)가 차지하였다(Fig. 2A, 2B and Table 2, 3, 4).
밀은 AA, BB, DD의 allohexaploid genome 구성을 하고 있다. GO annotation의 세 category (BP, CC, MF)에 대한 DEGs(총 1215개)의 염색체 분포를 확인한 결과, 모든 category 에서 2번 염색체(2A, 2B, 2D)에 가장 많은 DEGs가 분포한다는 것을 알 수 있었다. 반면 1번 염색체(1A, 1B, 1D)의 DEGs는 세 category에서 가장 적은 분포를 보였다. Genome 별로는 B (471), A (376), D (368) 순으로 많은 DEGs를 포함하고 있었으며 각각의 염색체 별 분포를 확인해보면 3B, 2B, 5B 순으로 많은 DEGs를 포함하고 있었다. 각 염색체 별 DEGs 분포는 Table 5에 나와 있다.


ABA 생합성 경로 관련 유전자
DEGs 분석을 위해 초반에 선발되었던 5119개의 유전자들 중 ABA 생합성 경로에 관여하는 유전자들을 분석하기 위해 애기장대에서 ABA 생합성에 관여하는 것으로 알려져 있는 유전자의 염기서열과 본 연구의 RNA-seq 결과 얻은 염기서열을 BLAST를 통해 비교하였다. ABA 생합성 경로 관련 유전자들에 염기서열이 매치된 유전자들을 확인한 결과, 매치된 ABA관련 유전자는 12개였으며 target 유전자와 매치된 밀의 reference 유전자는 34개로 확인되었으며, ABA 관련 유전자에 중복되는 reference 유전자를 제외하면 15개의 reference 유전자가 확인되었다(Table 6).
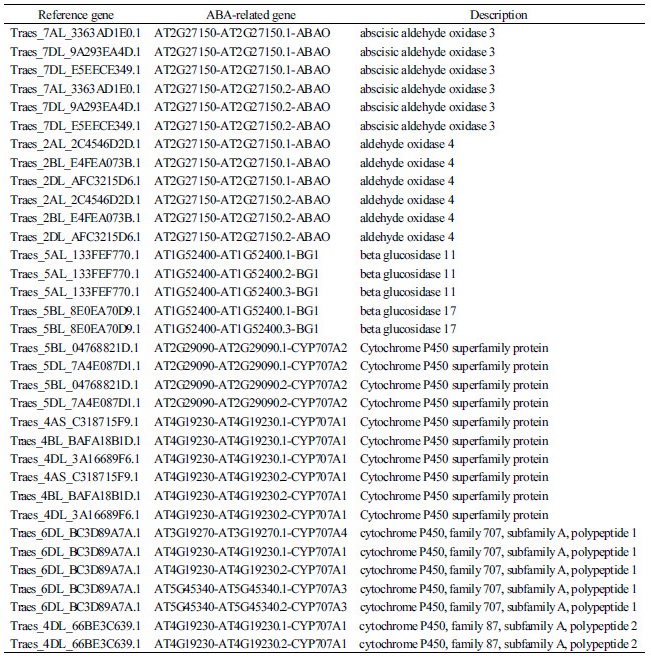
ABA 생합성 경로 관련 유전자들의 description을 확인해본 결과 본 실험에서는 크게 aldehyde oxidase (AAO), β glucosidase 및 cytochrome P450 (CYP707A)을 확인할 수 있었다. AAO는 ABA의 생합성 단계에서 작용하며 abscisic aldehyde를 ABA로 전환시켜주는 역할을 한다(Nambara et al., 2010). AAO는 또한 auxin (IAA)의 생합성에도 관련이 있으며 indole-3-acetaldehyde (IAD)를 IAA로 전환시켜주는 역할을 한다(Mano and Nemoto 2012). IAA는 ABA 및 ABA 신호전달 관련 유전자인 ABI3와 함께 상호작용하여 종자의 휴면과 발아를 조절하는 것으로 알려져 있다(Liu et al., 2013). 본 연구에서는 AAO3와 AAO4를 확인하였으며 각각의 유전자들은 7번 염색체와 2번 염색체(AAO3: 7AL, 7DL; AAO4: 2AL, 2BL, 2DL)에 위치하는 것을 알 수 있었다. 이들에 대한 발현을 분석해본 결과 AAO3는 control과 ABA 처리 샘플 사이에서 거의 발현에 차이를 보이지 않았으나 모든 AAO4는 up-regulation된 것을 확인하였다. AAO1-4 유전자들은 발현되는 부위가 특이적인 것으로 알려져 있으며 영향을 미치는 부위 및 기능에 차이가 있다(Seo et al., 2000). 본 실험에서는 유묘기 밀 식물체 전체를 샘플로 사용하여 AAO3과 AAO4의 부위별 발현 차이까지는 확인할 수 없었으나 추후 각 부위별 유전자들의 발현을 확인한다면 밀에서의 AAO3와 AAO4 유전자의 발현 위치 및 이 유전자들이 미치는 영향에 대한 연구가 될 것으로 사료된다. β glucosidase는 생물학적으로 비활성화 상태인 glucose-conjugated ABA (ABA-GE)를 가수분해 하여 활성화 상태인 ABA로 전환시켜주는 역할을 한다(Lee et al., 2006). 한편 cytochrome P450 (CYP707A)는 산화이화작용을 통해 ABA를 8’-hydroxy-abscisate로 전환시키며 최종적으로 phaseic acid (PA)로 전환시켜서 ABA를 불활성화 하는 역할을 한다(Saito et al., 2004; Zaharia et al., 2005). Liu et al. (2013)의 연구에서 후숙 과정을 거친 종자에 발아 및 ABA를 처리했을 경우 ABA 생합성 경로 관련 유전자들 중 xanthoxin을 ABA aldehyde로 전환시켜주는 ABA2 유전자가 가장 큰 발현 변화를 보였으나 본 연구의 RNA-seq 결과 상에는 ABA2를 확인할 수 없었다.
ABA는 ABA 수용체인 PYR/PYL/RCAR과 결합하여 ABA에 대한 반응을 유도하는 SnRK를 억제하는 PP2C가 작용하지 못하게 한다(Nakashima and Yamaguchi-Shinozaki 2013). ABA 생합성 경로와 함께 본 실험에서는 ABA 신호전달 과정에 관여하는 유전자들 약 25개(PYR/PYL/RCAR: 4, PP2C: 6, SnRK2: 13, ABF: 2)를 확인하였으나 일부의 SnRK2와 PP2C를 제외한 대부분의 유전자들은 control과 ABA처리 시 발현차이가 거의 없었다. 13개의 SnRK2 유전자 중 6개의 유전자가 up-regulation되었으며, 밀의 염색체 그룹 1과 2에 각각 3개씩(1AL, 1BL, 1DL, 2AL, 2BL, 2DL) 유전자가 존재하는 것을 확인하였다. Liu et al. (2013)는 ABA생합성 뿐 아니라 신호전달에 관련된 유전자들의 발현 변화에 대한 대량 분석을 microarray를 통해 수행하였으며, 이들에 따르면 ABA 수용체 PYR/PYL/ RCAR 및 일부 PP2C가 up-regulation 되었으며 본 실험 결과와는 다르게 SnRK2에서는 발현에 차이가 없는 것으로 나타났다. 이러한 차이는 품종 및 ABA에 대한 반응 민감도 차이 뿐 아니라 실험이 수행된 샘플의 부위 및 시기에 의한 것으로 판단된다.
본 실험에서는 대량전사체 분석을 통해 유묘기 밀에서 ABA 처리 시 차별적으로 up- 또는 down- regulation 되는 유전자 및 ABA 생합성 경로 관련 유전자들의 발현 및 정보를 확인하였다. 이를 통해 종자의 휴면과 발아 및 스트레스에 반응하는 유전자들에 대한 대량정보 및 단서를 확인할 수 있었으며, 추가적으로 다양한 수분스트레스 처리 및 그에 따른 전사체 발현 연구를 진행한다면 식물체 내에서의 ABA 작용 및 그와 관련된 전사체에 대한 심도있는 분석이 될 것으로 사료된다.
